|
Biodiversity Information Science and Standards : Conference Abstract
|
|
Corresponding author: Scott A Chamberlain (myrmecocystus@gmail.com)
Received: 06 Apr 2018 | Published: 21 May 2018
© 2018 Scott Chamberlain
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Chamberlain S (2018) Phylogeny Based Biodiversity Data Queries. Biodiversity Information Science and Standards 2: e25589. https://doi.org/10.3897/biss.2.25589
|
|
Abstract
There is a large amount of publicly available biodiversity data from many different data sources. When doing research, one ideally interacts with biodiversity data programmatically so their work is reproducible. The entry point to biodiversity data records is largely through taxonomic names, or common names in some cases (e.g., birds). However, many researchers have a phylogeny focused project, meaning taxonomic names are not the ideal interface to biodiversity data. Ideally, it would be simple to programmatically go from a phylogeny to biodiversity records through a phylogeny based query.
I'll discuss a new project 'phylodiv' (https://github.com/ropensci/phylodiv/) that attempts to facilitate phylogeny based biodiversity data collection (see Fig.
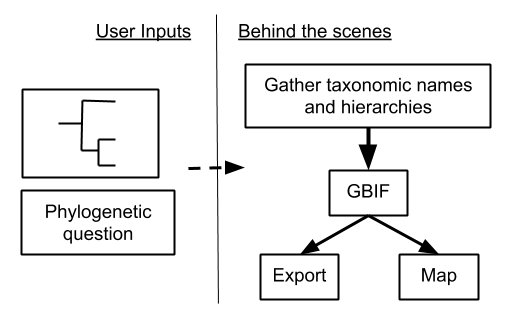
High level diagram of workflow for phylodiv. User inputs are a phylogeny and a phylogenetic question (e.g., A vs. sister of taxon A). "Behind the scenes" refers to the internal things that happen that the user doesn't have to worry about.
We already have R tools to do nearly all parts of the work-flow shown above: there's a large number of phylogeny tools, 'taxize'/'taxizedb' can handle taxonomic name collection, while 'rgbif' can handle interaction with GBIF, and there's many mapping options in R. There are a few areas that need work still however.
First, there's not yet a clear way to do a phylogeny based query. Ideally a user will be able to express a simple query like "taxon A vs. its sister group". That's simple to imagine, but to implement that in software is another thing.
Second, users ideally would like answers back - in this case a map of occurrences - relatively quickly to be able to iterate on their research work-flow. The most likely solution to this will be to use GBIF's map tile service to visualize binned occurrence data, but we'll need to explore this in detail to make sure it works.
Keywords
phylogeny, biodiversity, GBIF, R, programmatic
Presenting author
Scott A Chamberlain
Presented at
TDWG 2018 - Biodiversity Information Standards Meeting
Dunedin, New Zealand